
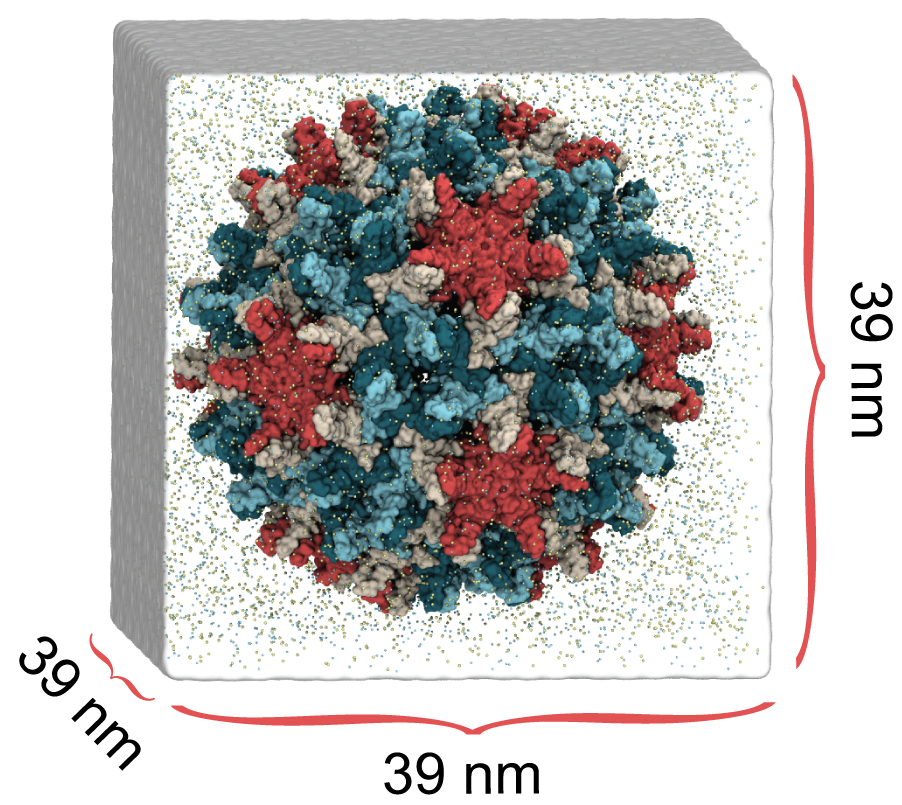
Coronavirus (SARS-CoV-2) Viral Particle

Structural components of SARS-CoV-2
In collaboration with the Perilla Lab at the University of Delaware, we are developing an atomistic computational model of the SARS-CoV-2 viral particle. SARS-CoV-2 is the virus that causes COVID-19. Constructing a model of this magnitude and complexity begins with modeling the individual structural components of the virus, including the lipid bilayer envelope, the proteins embedded in its surface, and the genome-containing nucleocapsid packed within its core. The spike (S) protein mediates adhesion and entry of the virus, the membrane (M) protein plays an essential role in assembly of the viral particle, and the envelope (E) protein forms a pentameteric ion channel called a viroporin. The nucleocpasid (N) protein, composed of N-terminal and C-terminal domains, forms a helical architecture to encase the viral RNA.
Hepatitis B Virus Capsid
Hepatitis B virus (HBV) causes severe liver disease. Although a vaccine to prevent HBV is available, there is no cure to treat people who are already infected. Researchers aim to design new drugs against HBV that target its capsid, a protein shell at the core of the virus that encloses its genetic blueprint. During infection, the capsid drives delivery of the blueprint to the host cell nucleus, where it is used to generate new copies of the virus. While the structure of the HBV capsid had been previously determined, its motion, which underlies its role in HBV infection, had not been characterized at the atomic level until recently.
HBV capsid simulation system, 6 M atoms
In a study published in eLife, we used molecular dynamics simulations on the microsecond timescale to observe the motion of the HBV capsid and its influence on surrounding water molecules and ions. We learned that the capsid is extremely flexible, and that this is likely the reason experimental microscopes have not been able to obtain high-resolution images of the capsid. We also learned that the capsid can distort asymmetrically, which may be important for the capsid to accommodate the asymmetric blueprint it encloses, or to squeeze through the nuclear pore into the host cell nucleus.
Finally, we learned that the capsid translocates ions across its surface through triangular-shaped pores, and that positively-charged ions translocate five times faster than negatively-charged ions. In the human body, the capsid proteins include positively-charged tails that expose themselves to the capsid surface, which signals the host cell to transport the capsid towards its nucleus. Our observations suggest that the tails are exposed through the capsid’s triangular pores, not through hexameric pores as previously thought, and that the capsid can control this exposure based on changes in tail charge that also occur during infection.
By revealing details about how the HBV capsid functions, our results imply new strategies to target the capsid with drugs. For example, drugs designed to rigidify the capsid could inhibit asymmetric distortion, and drugs designed to block triangular pores could inhibit cellular signaling. Importantly, our results also indicate that cryo-electron microscopy, which won the 2017 Nobel Prize in Chemistry, may be limited in reaching atomic resolution for biomolecules as flexible as the HBV capsid.
Virus Capsids as Drug Targets
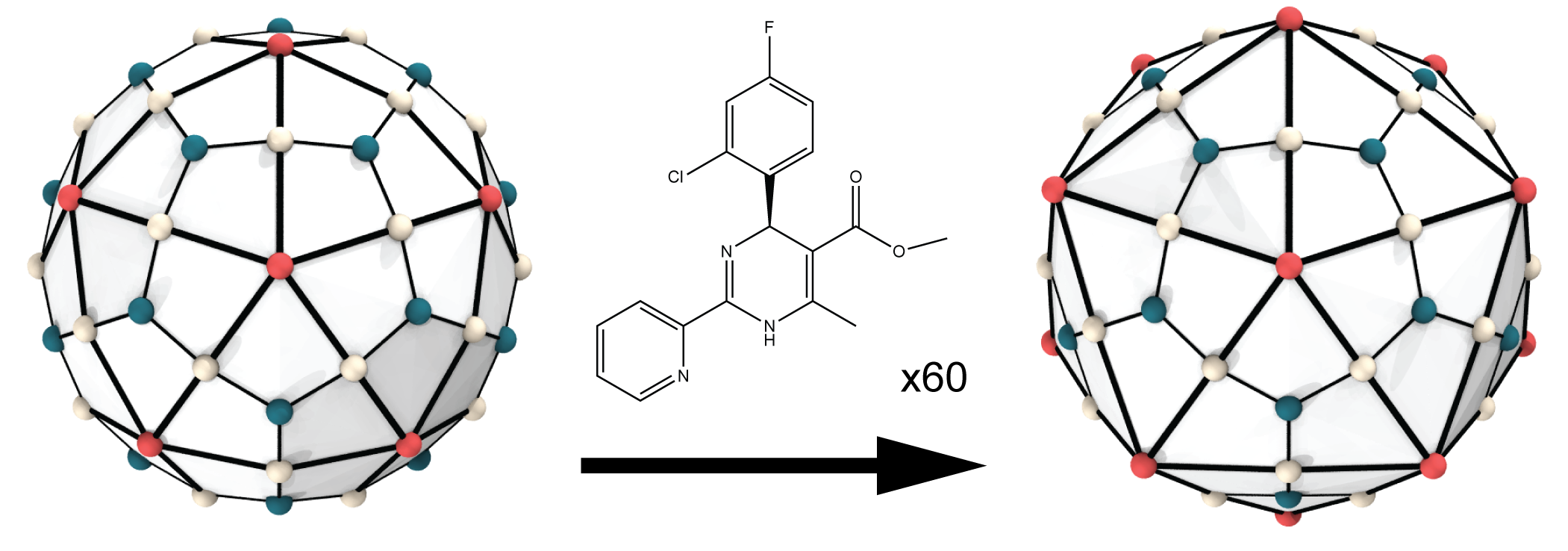
HAP1 drugs induce faceting of HBV capsid
In a study published in JPCL, we demonstrated that computational investigations of virus capsids as drug targets must be performed using full chemical detail. In other words, all of the atoms in the system must be represented explicitly. When all-atom molecular dynamics simulations were used, even subtle structural effects induced by drug molecules could be captured. For example, when the HBV capsid was simulated with 60 copies of the compound HAP1 bound, the capsid structure evolved to adopt a more faceted shape. This effect was subsequently observed by our collaborators in cryo-electron microscopy images of drug-bound HBV capsids.
Dynein Motor Domain

Dynein walks on microtubules
Cytoplasmic dynein is a motor protein that walks along microtubule tracks, towing cargo toward the center of the cell. Under normal circumstances, dynein cargo includes vesicles and organelles. In the case of infection, some viruses can hijack dynein to transport their genome-containing capsid to the cell nucleus.
Each dynein molecule contains two motor domains that are fueled by the hydrolysis of ATP to ADP+Pi. The energy released from the reaction powers each motor domain to undergo a mechanical cycle of unbinding from and rebinding to microtubules, i.e. stepping. The alternate stepping of two motor domains allows dynein to walk along microtubules, while towing cargo.
Despite the fundamental role of dynein in human biology and the relationship between dynein dysfunction and disease, dynein remains the least understood of all cytoskeletal motor proteins. Our research aims to investigate the mechanism and mechanical properties of dynein by integrating computing with experiment.
Dynein Mechanism

Anatomy of dynein motor domain
One long-standing question regarding the dynein mechanism is: Which structural component carries out the lever-action for the power-stroke that pulls cargo forward following a step, the stalk or the linker? Hypothetically, in a stalk-lever mechanism, the dynein ring would tilt dramatically relative to the microtubule during power-stroke; in a linker-lever mechanism, the dynein ring would maintain its orientation relative to the microtubule during power-stroke.
In a study published in PNAS, our collaborators tested this hypothesis using pol-TIRF microscopy to track ring tilting dynamics for dynein molecules walking in real time. Their results indicated that the dynein ring actually tilts only slightly relative to the microtubule during power-stroke, inconsistent with either model for the mechanism. We used all-atom molecular dynamics simulations on the multi-microsecond timescale to characterize the mechanical compliances of the dynein stalk and found that stalk flexing could account for the slight ring tilting observed in experiment, given a linker-lever mechanism.
Based on our combined findings, we propose that the dynein power-stroke is carried out by straightening of the linker, but depends also on the motion of the stalk. Besides pulling cargo forward, the power-stroke of the stepping motor domain pulls on the trailing motor domain, creating tension between them and causing their stalks to flex. When the trailing motor domain unbinds from the microtubule to take a step, the release of tension springs it ahead, biasing forward motion.
Tactile Visualization of Proteins
TactViz is our VMD plugin for tactile visualization of protein structures, designed to improve accessibility of structural biology and related fields for researchers who are blind or visually impaired. (a) 3D printed model of GB1. (b) variable-height tactile graphic of GB1 rendered with VMD+TactViz and printed using Pictures In A Flash machine. (c) variable-height tactile visualization of GB1 on Graphiti electronic refreshable tactile display, driven by VMD+TactViz. More information on TactViz is available through our publication in JSESD.

TactViz: A VMD Plugin for Tactile Visualization of Protein Structures
